Search results
Search for "quantum chemistry" in Full Text gives 25 result(s) in Beilstein Journal of Organic Chemistry.
Understanding the competing pathways leading to hydropyrene and isoelisabethatriene
Beilstein J. Org. Chem. 2022, 18, 972–978, doi:10.3762/bjoc.18.97

- potentially the roles specific active site residues play during catalysis [23][24][27]. Conclusion In the current work we performed gas-phase quantum chemistry calculations for the competing reaction pathways leading to IEs A and B as well as hydropyrene and hydropyrenol. In the former two reactions there is
- calculations and then chose the lowest energy conformer as representative of each carbocation intermediate in the mechanism. Hence, each intermediate state is represented by a single conformer, which is the lowest energy conformer found. Quantum chemistry calculations Optimizations and subsequent frequency





Lipophilicity trends upon fluorination of isopropyl, cyclopropyl and 3-oxetanyl groups
Beilstein J. Org. Chem. 2020, 16, 2141–2150, doi:10.3762/bjoc.16.182

- relates to the difference in Gibbs energy of the free ligand conformations of this solute in the respective solvents. Quantum chemistry calculations of Gibbs energies in octanol and water thus provide theoretical estimations of lipophilicities. Such estimations require systematic conformational analyses












Models of necessity
Beilstein J. Org. Chem. 2020, 16, 1649–1661, doi:10.3762/bjoc.16.137

- ; chemical structure representation; chemical structure models; language of chemistry; quantum chemistry; Introduction This article originally arose out of our discussions for the 2020 Beilstein Bozen Meeting “Models of Convenience” [1], which had to be postponed due to Covid-19 pandemic. It essentially
- 3D ball-and-sticks model only represents one of several possible conformations of clozapine but that all these conformations are inherent in the 2D-line structure. The dichotomy, really that between fundamental quantum chemistry and the common bonding models, which is made evident in Figure 1 and
- approximation for computational quantum chemistry has long attained the status of a bonding model to such an extent that many chemists outside the theoretical community are unaware that it is an approximation. Molecular-orbital bonding models based on the LCAO approximation discuss chemical bonds in terms of





In silico rationalisation of selectivity and reactivity in Pd-catalysed C–H activation reactions
Beilstein J. Org. Chem. 2020, 16, 1465–1475, doi:10.3762/bjoc.16.122
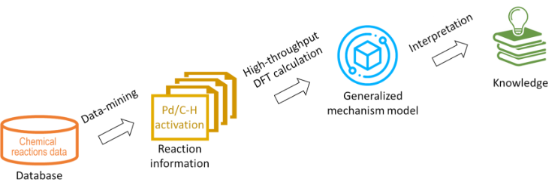
- , reaction conditions, and so on [23]. In order to achieve accurate and efficient reaction prediction, a mechanism-based method was chosen to direct quantum chemistry calculations and predictions, see Figure 1. For the Pd(II)-catalysed C–H activation reactions, there are two main commonly accepted mechanisms



Preparation of 2-phospholene oxides by the isomerization of 3-phospholene oxides
Beilstein J. Org. Chem. 2020, 16, 818–832, doi:10.3762/bjoc.16.75

- double bond migration pathways were elucidated by quantum chemical calculations. Keywords: chlorophosphonium salts; isomerization; 2-phospholene oxides; 3-phospholene oxides; quantum chemistry; Introduction P-Heterocyclic derivatives are valuable targets in synthetic organophosphorus chemistry [1][2][3







Azologization and repurposing of a hetero-stilbene-based kinase inhibitor: towards the design of photoswitchable sirtuin inhibitors
Beilstein J. Org. Chem. 2019, 15, 2170–2183, doi:10.3762/bjoc.15.214

- temperature. Computational details: All calculations were carried out using the TURBOMOLE version 7.2 quantum chemistry package [67]. Geometry optimizations of all compounds in different conformers were carried out using DFT with PBE approximation to the exchange-correlation (XC) functional and employing the










1,2,3-Triazolium macrocycles in supramolecular chemistry
Beilstein J. Org. Chem. 2019, 15, 2142–2155, doi:10.3762/bjoc.15.211

- also determined the dissociation constant pKa of this acid−base switchable MIM system by an indicator method which could make a quantitative and precise estimation of its dynamics with environmental acidity change. By combining NMR spectroscopic data and quantum chemistry calculations, a motion

















Archangelolide: A sesquiterpene lactone with immunobiological potential from Laserpitium archangelica
Beilstein J. Org. Chem. 2019, 15, 1933–1944, doi:10.3762/bjoc.15.189
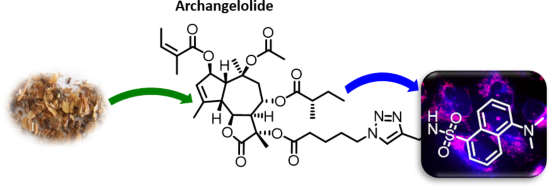
- structure of compound 2 was obtained by editing the DTB structure. First, we computed General Amber Force Field parameters for molecules of compound 1 and 2 in Antechamber software. Charges were calculated by restrained electrostatic potential method (RESP) based on a wave function calculated using quantum
- chemistry at the HF/6-31G*//HF/6-31G* level. Compound 1 molecule was manually docked into the SERCA binding site for thapsigargin using UCSF CHIMERA 1.10.2 (University of California, San Francisco) software by two approaches. In the first one, the molecule was docked so that the spatial orientation of the












The phenyl vinyl ether–methanol complex: a model system for quantum chemistry benchmarking
Beilstein J. Org. Chem. 2018, 14, 1642–1654, doi:10.3762/bjoc.14.140








Are dispersion corrections accurate outside equilibrium? A case study on benzene
Beilstein J. Org. Chem. 2018, 14, 1181–1191, doi:10.3762/bjoc.14.99

- of dispersion correction models. Note, this is different to seamless approaches like MP2, RPA or other quantum chemistry methods which include dispersion forces automatically. Common van der Waals corrections can be broadly divided into three categories, as will be detailed below. Substantial effort





Correlation effects and many-body interactions in water clusters
Beilstein J. Org. Chem. 2018, 14, 979–991, doi:10.3762/bjoc.14.83

- sound models of the interaction potential for water to be used in molecular simulations. In particular, the role of many-body interactions beyond the two-body interactions, which are often not explicitly taken into account by empirical force fields, can be accurately described by quantum chemistry
- electron correlation effects. Efficient quantum chemistry approaches for describing intermolecular interactions between water molecules may therefore describe higher-body interactions on an uncorrelated Hartree–Fock level without a serious loss in accuracy. Keywords: dispersion; many-body effects; water
- . These commonly depend on a number of empirical parameters that are determined either by a fit to experimentally known liquid or bulk properties, or by fitting to energies from ab initio quantum chemistry methods. The most popular potentials for water are the TIP3P [2], TIP4P [2][3] and TIP5P [4][5












An uracil-linked hydroxyflavone probe for the recognition of ATP
Beilstein J. Org. Chem. 2018, 14, 747–755, doi:10.3762/bjoc.14.63

- Marton Bojtar Peter Zoltan Janzso-Berend David Mester Dora Hessz Mihaly Kallay Miklos Kubinyi Istvan Bitter Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, Hungary MTA-BME Lendület Quantum Chemistry Research Group, Department of








Fluorescent nucleobase analogues for base–base FRET in nucleic acids: synthesis, photophysics and applications
Beilstein J. Org. Chem. 2018, 14, 114–129, doi:10.3762/bjoc.14.7

- of qA inside DNA disqualifies it for use as a base–base FRET donor. In order to maintain the base-analogue properties and achieve improved photophysical properties, we used quantum chemistry-supported design and developed a series of four, second generation, quadracyclic adenine analogues, qAN1–qAN4

















Complexation of molecular clips containing fragments of diphenylglycoluril and benzocrown ethers with paraquat and its derivatives
Beilstein J. Org. Chem. 2017, 13, 2056–2067, doi:10.3762/bjoc.13.203

- , Odesa 65080, Ukraine Department of X-ray Diffraction Studies and Quantum Chemistry, SSI ‘‘Institute for Single Crystals’’, National Academy of Sciences of Ukraine, Nauky Ave. 60, Kharkiv 61001, Ukraine Department of Inorganic Chemistry, V.N. Karazin Kharkiv National University, 4 Svobody Sq., 61122











Theoretical simulation of the infrared signature of mechanically stressed polymer solids
Beilstein J. Org. Chem. 2017, 13, 1710–1716, doi:10.3762/bjoc.13.165

- ][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74]. A variety of theoretical approaches have been developed to model external force using methods of quantum chemistry [9][75][76









New approach toward the synthesis of deuterated pyrazolo[1,5-a]pyridines and 1,2,4-triazolo[1,5-a]pyridines
Beilstein J. Org. Chem. 2017, 13, 800–805, doi:10.3762/bjoc.13.80

- position 3a to position 2 in cycloadduct 10 (path a) with the formation of intermediate 11 which, on further oxidation, gives product 8. Intermediate 11 is the most stable isomer among other dihydro intermediates according to quantum chemistry calculations by the M-06-2X 6-31G+(d,p) method (Figure 2) and






Creating molecular macrocycles for anion recognition
Beilstein J. Org. Chem. 2016, 12, 611–627, doi:10.3762/bjoc.12.60

- With accurate values for the 1:1 stabilities of various anion complexes deconvoluted from a veritable stew of other equilibria, quantum chemistry can be used to get an accurate picture of binding. First, we learn how to bring experiment and theory into agreement, then delve deeper into the origins of





















Solving the puzzling competition of the thermal C2–C6 vs Myers–Saito cyclization of enyne-carbodiimides
Beilstein J. Org. Chem. 2016, 12, 43–49, doi:10.3762/bjoc.12.6

- and Discussion The proper description of organic reactions involving diradical intermediates and transition state structures remains a difficult task in quantum chemistry [27][28][29][30][31][32][33][34][35]. Singlet diradicals have two electronic configurations that can only be treated accurately by







Efficient CO2 capture by tertiary amine-functionalized ionic liquids through Li+-stabilized zwitterionic adduct formation
Beilstein J. Org. Chem. 2014, 10, 1959–1966, doi:10.3762/bjoc.10.204

- the reaction mixture after CO2 absorption in the absence or presence of water. Influence of the ratio of LiNTf2/neutral ligands (PEG150MeTMG and PEG150MeBu2N) on the CO2 capacity of the coordinating mixtures. The quantum chemistry calculations (enthalpy changes) of the reaction between CO2 and







Host–guest complexes of mixed glycol-bipyridine cryptands: prediction of ion selectivity by quantum chemical calculations, part V
Beilstein J. Org. Chem. 2013, 9, 1252–1268, doi:10.3762/bjoc.9.142

- ; quantum chemistry; selective ion complexation; Introduction The present report continues a series of contributions from our group dealing with quantum chemical investigations of the selective complexation of alkali and alkaline-earth metal cations by supramolecular species, predominantly cryptands and




![[Graphic 2]](/bjoc/content/inline/1860-5397-9-142-i3.png?max-width=637&scale=0.295455) 2.2.bpy]+.
2.2.bpy]+.















Utilizing the σ-complex stability for quantifying reactivity in nucleophilic substitution of aromatic fluorides
Beilstein J. Org. Chem. 2013, 9, 791–799, doi:10.3762/bjoc.9.90

- accuracy since they do not take the relevant TS or solvation into account. Quantum chemistry methods exploring the PES on the other hand can, with sufficient modeling and appropriate level of theory, yield very accurate results and do not rely on predetermined experimental data. However, this procedure is






High-spin intermediates of the photolysis of 2,4,6-triazido-3-chloro-5-fluoropyridine
Beilstein J. Org. Chem. 2013, 9, 733–742, doi:10.3762/bjoc.9.83

- is based on comparison of their zero-field splitting (ZFS) parameters derived from experimental EPR spectra and calculated by quantum chemistry methods. The most accurate theoretical estimations are obtained at the PBE/DZ level of theory, which overestimates the experimental ZFS values of nitrenes by








New reactive intermediates in organic chemistry
Beilstein J. Org. Chem. 2013, 9, 613–614, doi:10.3762/bjoc.9.67

- exponential increase in computing power available to researchers, much of the mechanistic work nowadays is done using the tools of quantum chemistry. In particular, the various flavours of density functional theory have proven valuable in this respect, but high-level correlated methods such as CCSD(T) also
Regio- and stereoselective carbometallation reactions of N-alkynylamides and sulfonamides
Beilstein J. Org. Chem. 2013, 9, 526–532, doi:10.3762/bjoc.9.57

- Yury Minko Morgane Pasco Helena Chechik Ilan Marek The Mallat Family Organic Chemistry Laboratory, Schulich Faculty of Chemistry, and the Lise Meitner-Minerva Center for Computational Quantum Chemistry, Technion – Israel Institute of Technology, Haifa 32000, Israel 10.3762/bjoc.9.57 Abstract The






Recent advances in carbocupration of α-heterosubstituted alkynes
Beilstein J. Org. Chem. 2010, 6, No. 77, doi:10.3762/bjoc.6.77

- Ahmad Basheer Ilan Marek The Mallat Family Laboratory of Organic Chemistry, Schulich Faculty of Chemistry and the Lise Meitner-Minerva Center for Computational Quantum Chemistry, Technion-Israel Institute of Technology, Haifa 32000, Israel 10.3762/bjoc.6.77 Abstract Carbocupration of α
























